
Why AI Biology Needs Cell-Cell Interaction Data
July 8, 2026
The race to build the “ChatGPT of biology” is accelerating.
Across academia, biotech, and pharma, AI foundation models are being trained on massive collections of single-cell transcriptomic profiles with the goal of learning the rules of life from data. The long-term vision is ambitious: models that can simulate cellular behavior, predict responses to perturbation, and eventually support virtual cells capable of representing biology across molecular, cellular, and tissue scales. (Bunne et al., 2024)
If these models work, the way we study disease and design therapies could start to change. Researchers could simulate how cells respond to inflammation, infection, cancer, aging, or treatment before running every experiment at the bench. Instead of testing large numbers of candidates and learning slowly from what survives each experimental screen, scientists could use models to prioritize the drugs, antibodies, genetic perturbations, or engineered therapies most likely to shift biology in the desired direction. The best models would not replace experiments, but they could help researchers choose better experiments, spot likely failures earlier, and design improved medicines faster.
But as the field scales model size, compute, and training corpora, a more fundamental problem is becoming clear: most training data still captures cells as individual units.
That leaves out one of biology’s most important dimensions: what happens when cells encounter one another.
Biology is not only encoded inside cells. It emerges between them. To build models that better reflect living systems, AI biology will need cell-cell interaction data that captures how cells behave together.
More Single-Cell Data Is Not the Same as Better Training Data
Single-cell biology has increasingly followed the scaling logic of machine learning: larger datasets, larger models, and broader training corpora. Single-cell foundation models have expanded rapidly, moving from early studies with 1 million cells to newer atlases with more than 100 million cells. In practice, these models are being used to ask whether a cell’s molecular profile can reveal what state it is in and how it might respond to a drug, genetic change, or other perturbation. (DenAdel et al., 2026; Theodoris et al., 2023; Cui et al., 2024; Zeng et al., 2025)
But large-scale benchmarking suggests that single-cell foundation models do not automatically improve simply by adding more pretraining data. Performance gains can plateau depending on the task, model architecture, and dataset composition, while perturbation-prediction benchmarks have found that deep-learning models do not yet consistently outperform simple baselines across key evaluation settings. (DenAdel et al., 2026; Ahlmann-Eltze et al., 2025; Csendes et al., 2025; Wu et al., 2025)
The bottleneck is not only scale. It is signal.
Why Do Standard Single-Cell Approaches Miss Cell-Cell Interaction Signals?
Cell-cell interaction signals are often difficult to capture with standard single-cell approaches. Modern single-cell sequencing has transformed biology by making it possible to profile thousands to millions of cells at once. But many workflows still begin by dissociating tissues, tumors, organoids, or co-cultures into individual cells. In doing so, they can separate cells from the physical contacts and local signals that shaped their state, such as immune recognition of tumor cells or antigen presentation to lymphocytes. Spatial methods preserve tissue organization, but they still may not resolve which neighboring cell, timing window, long-range cue, or reciprocal interaction drove a specific response. (Di Carlo et al., 2026)
In other words, we often sequence the actors after the scene has ended. The resulting data can show what each cell looked like in that single frame, but not always what caused that state or whether the inferred interaction produced a functional outcome. A T cell activation signature may reflect direct tumor recognition, cytokine exposure, or bystander inflammation; a ligand-receptor pair may suggest communication without proving signaling occurred; and a cytotoxic gene signature does not prove target-cell killing.
For AI biology, this means models may be asked to predict multicellular behavior without direct examples of the interactions that produced it.
How Nanovials Turn Cell-Cell Interactions Into AI-Ready Data
To capture that missing interaction signal, researchers need experimental systems that keep defined cell partners together long enough to measure what happens between them.
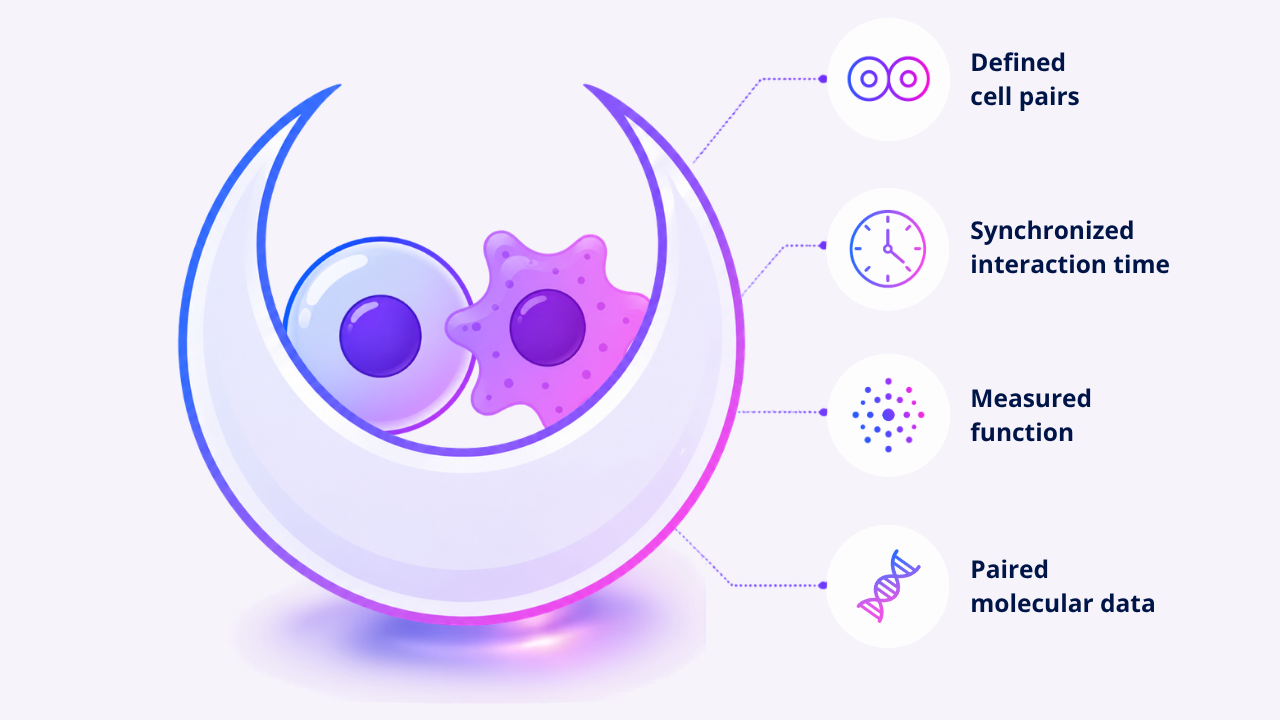
Nanovials provide a physical way to capture interaction context in defined cell pairs. These cavity-containing hydrogel microparticles create nanoliter-scale compartments around cells. In the Cell-Cell-seq workflow, Nanovials bring defined cell partners together, synchronizing the start of their interaction, measuring functional readouts, and connecting those paired interactions to downstream single-cell transcriptomic analysis while helping preserve fragile cell-cell contacts during handling and sorting. Researchers can not only ask, “What is this cell?” but also, “What happened when this cell met that cell?” (Baghdasarian et al., 2026)
That timing control is important. In bulk co-cultures, cell-cell contacts may begin at different times, involve different partner combinations, and become averaged across the population. Nanovials make it possible to compare defined interactions within a more controlled time window, enabling more consistent measurement of early and dynamic interaction responses.
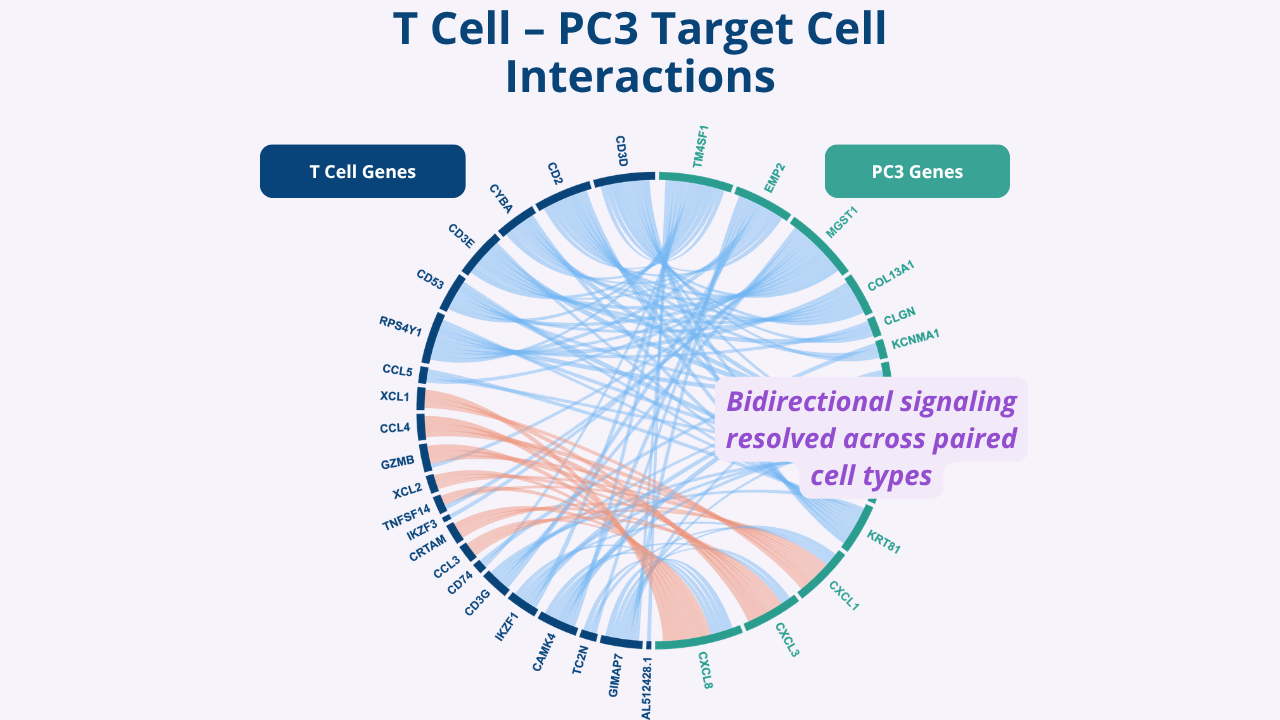
In antigen-matched tumor-T cell pairs, Cell-Cell-seq revealed coordinated transcriptional programs that were not simply the sum of the two cells in isolation, including contact-associated activation programs, partner-dependent signaling patterns, and heterogeneity across individual interactions. (Baghdasarian et al., 2026)
A related NK cell-tumor study revealed that Nanovial-measured activated cell pairs were harder for single-cell atlas-trained foundation models to reconstruct than held-out singlets, highlighting the gap between current single-cell training data and contact-dependent interaction states. (Liang et al., 2026)
Together, these studies show how Nanovial-enabled workflows can turn cell-cell interactions into measurable, sortable, and sequence-ready examples of partner-specific biology.
What Cell-Cell Interaction Data Could Teach Foundation Models
The next generation of biological AI will need to learn more than cell identity or cell state. It will need to learn how cellular behavior changes with partner identity, timing, perturbation, and local context. (Di Carlo et al., 2026)
That kind of learning is essential for understanding biology in living systems. Cancer, autoimmunity, fibrosis, infection, and tissue repair are shaped by how cells communicate, compete, suppress, activate, and remodel one another.
If future AI models are expected to predict these processes, they will need training data that captures cells in context, in contact, and in action.
Nanovials are helping make those interaction-resolved datasets possible.
Learn more about how Nanovials generate interaction-resolved datasets for functional biology.
References
Ahlmann-Eltze, C., Huber, W., & Anders, S. Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines. Nature Methods, 2025.
Baghdasarian, S., Wang, Q., Langerman, J., Mao, Z., Wright, H., Gee, C., Cheng, D., McLaughlin, J., Lee, J. K., Chen, X., Garcia, K. C., Li, J. J., Witte, O. N., Plath, K., & Di Carlo, D. Systematic mapping of emergent transcriptional states in interacting single-cell dyads by Cell-Cell-seq. bioRxiv, 2026.
Bunne, C., et al. How to build the virtual cell with artificial intelligence: Priorities and opportunities. Cell, 2024.
Csendes, G., Sanz, G., Szalay, K. Z., & Szalai, B. Benchmarking foundation cell models for post-perturbation RNA-seq prediction. BMC Genomics, 2025.
Cui, H., et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 2024.
DenAdel, A., Hughes, M., Thoutam, A., Gupta, A., Navia, A. W., Fusi, N., Raghavan, S., Winter, P. S., Amini, A. P., & Crawford, L. Evaluating the role of pretraining dataset size and diversity on single-cell foundation model performance. Nature Methods, 2026.
Di Carlo, D., Morsut, L., McCain, M. L., Wright, H. J., Abedi, M., Yamada-Hunter, S. A., Zhang, J. Z., Backus, K., Chung, E., Wang, Y., Rando, T. A., Cai, L., Thomson, M., Elowitz, M. B., Lee, J. K., & Witte, O. Mapping and engineering the human cell-cell interactome. Nature Biotechnology, 2026.
Liang, J., Shao, D., Di Carlo, D., & de Rutte, J. M. Cell-Cell-Seq resolves contact-associated NK cell activation in defined tumor cell dyads. bioRxiv, 2026.
Theodoris, C. V., et al. Transfer learning enables predictions in network biology. Nature, 2023.
Wu, Y., Wershof, E., Schmon, S. M., Nassar, M., Osiński, B., Eksi, R., Yan, Z., Stark, R., Zhang, K., & Graepel, T. PerturBench: Benchmarking Machine Learning Models for Cellular Perturbation Analysis. NeurIPS, 2025.
Zeng, Y., et al. CellFM: a large-scale foundation model pre-trained on transcriptomics of 100 million human cells. Nature Communications, 2025.